From “Method Development, Optimization and Trouble Shooting for High Performance Capillary Electrophoresis”
There are several aspects of precision that are very critical to success in Capillary Electrophoresis.
Quick Links to Topics on this Page:
To control these aspects the following variables must be controlled to the best of your ability:
- Injection Size
- Temperature
- Viscosity of samples
- Samples Ionic Strength
- Peak Detection and Integration
- Sample Evaporation
This chapter deals with each of these parameters within the following topics.
Quick Links to Topics on this Page:
-
THE CAPILLARY SURFACE
-
THE BACKGROUND ELECTROLYTE (BGE)
-
INJECTION PRECISION
-
HYDRODYNAMIC INJECTION PRIMER
-
ELECTROKINETIC INJECTION PRIMER
-
STACKING AND TRACE ENRICHMENT TECHNIQUES
-
SAMPLE PREPARATION TECHNIQUES
Sources of Migration Time Variation
| Source | Remedy |
| Wall Effects |
|
| Sample Overload | Dilute the Sample |
| Buffer Depletion |
|
| Temperature |
|
| Sample Ionic Strength |
|
| Inappropriate pH | Use Mobility Plot to Optimize pH |
| Buffer Problems |
|
Sources of Peak Area and Resolution Variances
| Source | Remedy |
| Injection |
|
| Sample Evaporation | Use Sample Container Closure that Seals Well |
| Sample Depletion | Use Fresh Sample for Electrokinetic Injection |
| Integration Parameters | Adjust Integration Parameters to HPCE Quality |
| Migration Time Variance | See Above Chart |
| Buffer Depletion | See Above Chart |
THE CAPILLARY SURFACE
The bare fused silica capillary represents a “weak link” in HPCE separations reproducibility due to inherent chemical nature. Generation of the Electro Osmotic Flow (EOF) is generated in part by the capillary surface’s interaction with the BGE. It is not uncommon to see un-controlled capillary surface-BGE conditions creating variability on a run-to-run, day to day and capillary-to-capillary basis.
Capillaries from different manufacturer’s and even from the same manufacturer could have enough variability in it that a reproducible separation is impossible without intervention by the scientist.
It should also be noted that different manufacturer’s capillaries would vary in their UV transparency as well.
Inter-Sample Wash Procedures
To maintain a pristine and reproducible capillary surface, it is best accomplished by
- Not injecting proteinaceous material into the capillary,
- Using additives that minimize binding to the capillary wall and
- Performing inter-sample washes.
A typical inter-sample wash uses either 1.0N or 0.1N sodium hydroxide for a few minutes followed by re-equilibration of the capillary surface with BGE for several minutes. For proteinaceous materials that have been injected into the capillary, it is often recommended that SDS be used to remove the proteins.
There are other more advanced wash procedures for more difficult situations when broadened peaks appear or there is a noisy baseline after a good run.
Advanced Column Wash Procedure
| A. 1.0 N Hydrochloric Acid | 2 minutes |
| B. 0.1 N Sodium Hydroxide | 5 minutes |
| C. CEwater | 2 minutes |
| D. BGE | 5 minutes (before injection) |
To store your capillary, it is suggested that you perform the advance wash procedure and then leave the capillary filled with 0.1N NaOH or dried out with Nitrogen.
Coated Capillaries
Coated capillaries may help resolve wall effects but are not immune to adsorption. Another factor is that the coating may not be stable and is most likely not permanent. Coated capillaries can be washed the same as uncoated but it is best to check with the manufacturer of the capillary before using the Advanced Wash Procedure. Usually coated capillaries can be washed with a 10-fold concentration of BGE.
Neutral Markers and Internal Standards To Determine Capillary Usefulness
You can monitor your capillary condition or “health” by adding neutral markers or internal standards into each run. The neutral markers serves as a measure of the EOF and you can determine its reproducibility between runs. Mesityl Oxide and Benzyl Alcohol are routinely used for CZE; add a marker in a 0.5% concentration to the sample before injection. For MEKC or MECC you cannot use Benzyl Alcohol due to the fact that it will interact with the micelles.
At low pH, a neutral marker may have a very, very slow EOF and may not elute at all.
Viscous BGE or capillaries coated to produce very slow EOF will have the same effect that low pH CZE does.
The recommended qualitative reporting parameter for determining the quality of your CZE is the neutral marker with “corrected migration times”. This will correct for the fluctuations in migration times that may occur. When EOF variation becomes too high on a run-to-run basis, it is recommended that the source of variation be determined and controlled. The use of the neutral marker is best used to determine if the capillary has surpassed its useful lifetime.
Factors Controlling EOF
EOF, the scientist’s friend and foe; while permitting the simultaneous separation of anions, cations and neutral species, controlling this vital parameter can be very difficult. Since the EOF is controlled by so many parameters, it is recommended that as many parameters as possible be held constant to obtain stable EOF. These parameters include:
- Capillary Condition
- Field Strength
- pH
- Concentration and Conductivity of BGE
- Additives to the BGE
- Viscosity of BGE
- Temperature
Even when coated capillaries are use, the capillary condition is still unknown and difficult to control so it is recommended that you ensure all other variables are very carefully controlled.
When using bare fused silica capillaries, EOF usually varies from 10-5 cm2/Vs at pH to 10-4 cm2/Vs at pH 8. This means that the EOF increases between pH 3 and 8 by a factor of 10. If you chose to use a pH between 3 and 8, you can expect that any inadvertent changes to pH will cause substantial changes in EOF and therefore variation in migration time. From our standpoint of precision, pH of 2.5 is the best place to work when using simple BGE.
Coated capillaries have been used to suppress the EOF but these coatings become unstable at high alkaline pH.
Methylcellulose derivatives reduce the EOF but there is not much work done in CZE with it.
Linear alcohols such as methanol, ethanol and propanol reduce the EOF.
Cationic surfactants not only reduce the EOF they can reverse it.
It is important to remember that the EOF is reduced in proportion to the square root of the buffer (BGE) concentration and is inversely proportional to the buffer viscosity. Since the EOF is a function of the BGE viscosity, the capillary temperature must be controlled very tightly.
THE BACKGROUND ELECTROLYTE (BGE)
The preparation and use of the BGE can have a very surprising impact on precision. The problems encountered usually are:
- Failure to control ionic strength/Conductivity
- Stability/Bacterial Growth
- Buffer Depletion
Ionic Strength and Conductivity
The ionic strength and conductivity of the Background Electrolyte is a critical factor in achieving precision in HPCE. Both the mobility and the EOF are inversely proportional to the square root of the buffer concentration. If your BGE varies from day to day, you may see dramatic changes in your migration times.
Conductivity should be measured each day as part of your routine lab procedures and should be included in all of your standard operating procedures. Also, you should include the specific amount of the counter ion (i.e. lithium, sodium) in your method. This is a critical point in inter laboratory reproducibility. If different counter ions are selected, the migration times and results will most likely vary.
Stability of your Background Electrolyte (Buffers)
Some buffers are excellent media for bacterial growth. To minimize this, you should keep your buffers (BGE) in a refrigerator. To reduce contamination by bacteria, you can add a 3mM solution of Sodium Azide. Always bring buffers to room temperature before using them.
It is also recommended that before you use the BGE solution (buffer) that you filter it through a .2m syringe filter.
Buffer Depletion (Avoid Electrolysis)
Buffer ions move in two different directions.
Anions move toward the anode and Cations move toward the cathode. The catholyte (negatively charged electrode buffer) will eventually become depleted of anions and the anolyte will become depleted of cations. The end result of this will be electrolysis and the production of protons and hydroxyl groups resulting in a rise in pH in the catholyte and a fall in pH in the anolyte.
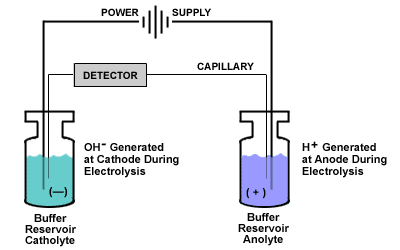
Two different pH environments are created and EOF will vary as protonated anolyte moves through the capillary.
Due to the pH differences in runs, resolution will therefore change along with migration times. You can reduce this cause of poor precision by using an adequate buffer capacity.
Another hint that will help reduce buffer depletion is to use large buffer containers and of course, you should change buffers (BGE) often.
Temperature
The Background Electrolyte’s (BGE) viscosity decreases as its temperature increases and therefore the frictional drag experienced by a migrating solute decreases as well. A rule of thumb is that for every 2º C of temperature increase you will see a corresponding increase in mobility. To achieve precision, you must have good temperature control of your capillaries.
INJECTION PRECISION
For new and inexperienced users of HPCE, the injection procedure is a good way to destroy your precision. You must develop command of the injection procedures: Hydrodynamic (pressure or vacuum) or Electrokinetic. Although both procedures have advantages and disadvantages, it is usually preferred to perform hydrodynamic injections.
Thinking in terms of electrophoresis and not chromatography, one must consider how the ions move about in a fluid solution under an applied electric field to develop a good understanding of the injection procedure and pitfalls it may have.
In the CE Primer’s Helpful Hints section, you can find “Trouble Shooting Common Injection Problems” to help with problems you may encounter.
Performing the Injection
When performing the injection procedure, a few factors must be considered. Injection of your sample is performed by placing the capillary into the sample vial and applying either pressure/vacuum or voltage. When the capillary is in the sample vial, solutes will diffuse into the capillary and BGE or buffer ions diffuse into the sample vial. To achieve precision, this potential for quantitative error and sample contamination, the separation should proceed within 1-3 seconds from inserting the capillary into the sample vial. Once the capillary is returned to the anolyte (buffer reservoir), it is important that the separation be started immediately.
Voltage Preconditioning
It has been reported that voltage preconditioning of the capillary will improve your migration time precision. A short procedure on how to do this is:
- Fill the Capillary/Column with fresh BGE
- Run only buffer through the Capillary/Column at 30kV for 2 minutes
- Inject your sample as described above.
Injection is done with voltage in the off position although it is probably better to inject with the voltage on but no commercial instrument has this feature.
Insulating the Sample During Injection Procedures
To develop the most precise HPCE method possible, it is important to consider all contributions to error including the sample moving back out of the capillary when voltage is applied. This occurs when BGE expands due to heating and this expansion is faster and greater than the rate of solute electrophoretic migration to the cathode.
This can be avoided with one of two procedures:
-
Voltage Ramping:
Voltage Ramping is when the voltage is incrementally increased giving the solute enough time to migrate far enough into the capillary to negate sample loss. -
Sample Insulation:
Sample Insulation is when you make a second injection of sample before you apply the voltage. This will push the solute far enough into the capillary to overcome the effects of thermal expansion of the BGE making it not significant.
HYDRODYNAMIC INJECTION PRIMER
Under most situations, hydrodynamic injection is the preferred means of injection in HPCE. Since a plug of material is introduced into the capillary/column, this form of injection is non-discriminatory. It will not change the sample concentration or matrix.
The equation governing the amount of material the is injected during hydrodynamic injection is:

It is significant to point out that the flow rate is proportional to the fourth power of the capillary diameter. If you change capillary sizes you must adjust the injection times in proportion to the square of the capillary diameter.
If you do not do this you will NOT get an identical injection plug length which is needed for precision. A correlation exists between the capillary diameter, injection volume and injection plug length. The flow rate is inversely proportional to the length of the capillary so the time must be adjusted accordingly.
Also, note that the viscosity of the both the BGE (buffer) and the sample will impact the amount of material actually injected. Internal standards are very useful to control some of these variables. Despite these problems, at least we are reasonably certain that the proper number of ions are introduced into the capillary.
If the ionic strength varies from sample to sample, the amount of peak compression (Stacking) that occurs will vary. This is very troublesome when the sample is of high ionic strength. Sample preparation may be required to place the solute in an HPCE “friendly” matrix.
ELECTROKINETIC INJECTION PRIMER
When you need to inject a very small amount of material, it is easily accomplished by using Electrokinetic injection. The equation that defines the amount of material injected by Electrokinetic injection is:

Electrokinetic injection must be used over hydrodynamic injection when the capillaries are very small or if the BGE or buffer is very viscous.
It has been reported that one of the greatest advantages of this type of injection is TRACE ENRICHMENT. When the EOF is low, it is possible to inject only ions into the capillary and substantial trace enrichment can occur. The problem with this technique is control and precision.
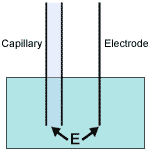
Electrokinetic injection. The field strength at the point of injection determines the number of ions that enter the capillary.
There are two bias problems associated with Electrokinetic Injection:
- Bias due to the conductivity of the sample
- Bias due to mobility differences among solutes (Sample Depletion)
The first bias is the most problematic. As the above figure indicates, the field strength at the point of injection is related to the conductivity of the sample. As sample total conductivity increases, the electric field strength falls off; fewer ions are injected.
A calibration curve will indicate a negative deviation from linearity at higher sample concentrations levels due to this bias. Making things even more problematic is that if the sample matrix contains ions other than the solute of interest (impurities, degradation products etc.) the matrix ions may migrate into the capillary effectively displacing solute ions.
The second bias, discrimination among solutes in the sample is also a problem for Electrokinetic injection. Highly mobile solutes are preferentially injected compared to lower mobility ions. Since mobilities can be measured, internals standards can be used with relative success. If the sample matrix is constant, external standards can be used to compensate for this bias.
Be aware that repeated injections from the same sample vial can result in sample depletion. If you note a gradual and continual decrease in peak height and area, then you may be experiencing sample depletion. This is unique to Electrokinetic injection.
STACKING AND TRACE ENRICHMENT INJECTION TECHNIQUES
Sample stacking, peak compression or Trace Enrichment can be accomplished on-line if the sample is injected in a low conductivity solution. When this occurs, the field strength in the injection zone is high relative to the BGE. As the frontal cations migrate toward the cathode, they cross the boundary at the front of the injection zone and enter the BGE.
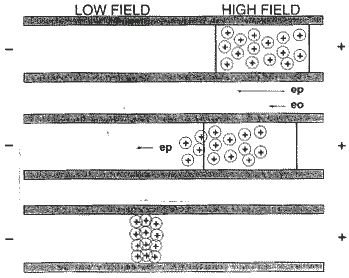
Mechanism for ionic strength mediated stacking
At the point when the ions move into the BGE, the field strength declines (the BGE has a different conductivity) and as a result, the ions slow down. The ions that are still in the injection zone are still moving at a fast speed. As a result, you get substantial peak (analyte) compression as in the third line of the drawing above. Your capillary can be filled with 5-10% sample with little to modest band broadening using this technique with good precision.
It is not recommended to attempt peak compression using water as the sample solvent/buffer with Hydrodynamic Injection. Although the field strength would be very high and theoretically you would expect to achieve peak compression, this is not optimal. The problem is that in reality, water injections lead to band broadening and the excessive heat generated over the injection plug can lead to decomposition of labile solutes.
If you are going to use Electrokinetic Stacking, it is recommended that you use a water PRIOR to injection of your sample. This will maintain a high field strength at the point of injection partially compensating for ionic strength differences between solutes as well as mobility discrimination. Usually, as the higher mobility solutes enter the capillary, the field strength drops causing further discrimination in favor of the lower mobility solutes.
pH Mediated Stacking for Zwitterionic Samples
For zwitterions, pH Mediated Stacking is very useful and can improve your precision. Your injection plug (sample solvent/Buffer) is a higher pH than the BGE/run buffer as shown in the drawing below.
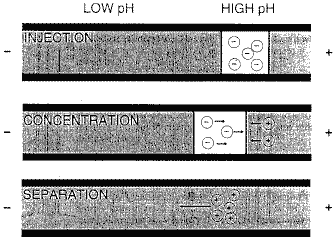
Mechanism for pH mediated stacking
In the first line of the drawing above, the sample which is dissolved in 10mM ammonium hydroxide is injected into the capillary. The BGE in front of the injection plug is 20mM Citrate, pH 2.5.
As the anionic solute migrates toward the anode, it crosses the barrier out of the injection solvent and into the BGE. The zwitterions flips its charge (line two of the above drawing) and reverses it direction of migration. Solute ions still in the ammonium hydroxide solution still migrate toward the anode.
As all the solute now flips its charge and is in the BGE, substantial peak compression or sample enrichment can occur. With this technique as much as 15% of the capillary volume can be injected without band broadening and with good precision.
Transient Isotachophoresis, The Ultimate Stacking Technique
Detailed discussion of this technique is beyond a CE Primer but it is important to understand what it can do for you. The benefit of this technique is that under optimal conditions, the capillary can be filled to 50% of its volume with no band broadening and with good precision. It is recommended that if this technique can be of use to you, contact an expert in this field that has experience.
Sample Carry Over in the Injection Process
Sample Carry Over is the contamination of a sample by the sample injected right before this one. This can become significant when 1.) viscous samples are analyzed or 2.) a high concentration sample is followed by a low concentration sample or a blank.
It is recommended that all methods are tested for carry over by measuring a high concentration sample (1mg/ml for example) followed by a blank injection. If significant carry over is found, the capillary and electrode assemblies should be automatically washed prior to starting the next run. See the washing procedure listed above. It is strongly recommended to perform inter sample washing to increase precision and increase the life of the capillary.
The Ubiquitous Injection, small but unavoidable
The smallest amount of material that one can inject is defined as The Ubiquitous Injection. What this really means is that if one simply immerses a capillary into a solution, a small amount of material will enter the capillary. This is why you should avoid very small injections. This will compromise the precision of the injection. It is recommended to not make injections for less than 0.5 seconds.
Injection Calibration; Rates of Injection
Measuring the actual amount of sample entering the capillary is not straightforward. We have defined equations that describe both Electrokinetic and Hydrodynamic injection, we must rely on internal and external standards to calibrate our HPCE system and our methods.
If you actually wish to measure the amount of material being injected into the capillary there are two procedures to follow to determine the “rates of injection”:
Rate of Injection (mm/s) = Capillary Length (mm) Time to Detector (s)
Inject a dye into the capillary and under magnification, examine how far the dye has moved into the capillary. The rate is defined similarly to the above equation by substituting the Capillary Length with the distance observed and the time to detector with time it takes to get to the place you measure (recommended the detection window). This only works with Hydrodynamic Injection and should not be used with Electrokinetic Injection.
SAMPLE PREPARATION TECHNIQUES AND PRECISION
Not all samples will allow you to “dilute and shoot”. Complex sample matrices or when trace analysis is required, good sample preparation techniques can make the difference between precise results and variable results.
Often, good sample preparation techniques will:
- Improve Capillary Lifetime
- Reduce Wall Effects
- Improve Selectivity
- Improve Precision and Accuracy
- Improve Sensitivity
Improving the (1.) capillary lifetime and (2.) reducing wall effects can be achieved by removing materials in the sample matrix that can adhere to the capillary walls. Usually proteins found in biological fluids will contaminate the capillary if they are not removed. If you are doing MECC or MEKC this step may be avoided because the surfactants used will solubilize proteinaceous materials.
Anything that adheres to the capillary walls will effect (3.) Selectivity and (4.) Precision and Accuracy as well. These two points are inter related, if Selectivity is high, Precision and Accuracy is usually good as well.
Using off line enrichment techniques such as Solid Phase Extraction (SPE) often improves sensitivity (5.) and also many materials that adhere to capillary walls are removed. Often direct injection is possible but just as often, off line enrichment techniques improves the five points made above.
Other off line sample preparation methods that can be used include but are not limited to:
- Filtration through a .2µ syringe filter
- Dialysis (Desalting)
- Solid Phase Extraction
- Centrifugation
- Protein Precipitation with Acetonitrile
- Affinity CE/CEC
Sample Evaporation
It is important to take notice of your instruments autosampler or sample vials. Many instruments do not offer completely sealed vials. Even though it may appear that this is not a critical factor for your specific sample, it is important to remember that working with such very small samples, even slight evaporation can be significant.
If your samples are dissolved in organic solvents, this becomes critical. While internal standards help to solve some of the quantitative problems due to evaporation, it is still recommended that you use the appropriate sample containers. If your instrument’s autosampler can be cooled, this will help to minimize this problem.
Peak Generators and Data Systems
Due to the very narrow peaks of HPCE, it is important that your data system has a data collection device that is compatible with HPCE. The sampling rates of typical HPLC integrators and data systems are too slow for CE. Using the wrong device could result in under sampling of data. This will create errors since peak start, maximum and stop may be missed or improperly averaged. If you are using a home built CE system this may be important. If you are using a commercially available CE Instrument, this will most likely not be your problem.